Geometric Deep Learning on Protein Design
Supervisor: Prof. Stefano Angioletti-Uberti
Protein Folding | Deep Neural Networks | Curse of Dimensionality | Representation Learning
Building Rotation Invariant Representation for Inverse Protein Folding Using Spherical Harmonics
List of Contents
- Protein Folding
- Protein Structure Determination
- Traditional and Deep Learning Approaches in Protein Design
- Train of Thought
Protein Folding
Proteins are complex biomolecules that exhibit a wide range of structural and functional diversity due to their evolution over billions of years. The primary structure of a protein is its amino acid sequence, the secondary structure is the way in which the protein chain folds into specific shapes, and the tertiary structure is the overall three-dimensional shape of the protein.
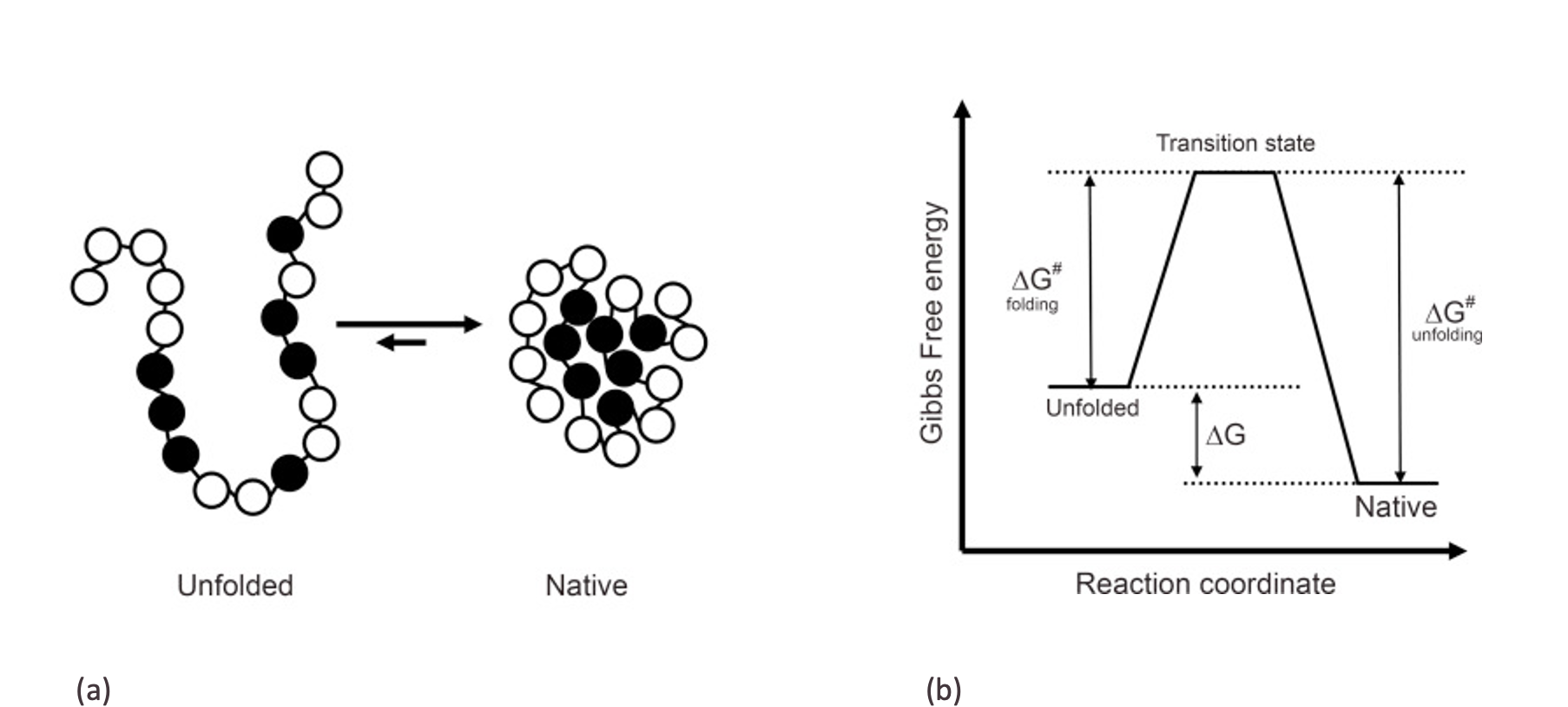
Protein folding is the process by which a protein molecule adopts its specific, biologically active three-dimensional structure from a random coil. Proteins are made up of long chains of amino acids that are joined together by peptide bonds. When a protein is synthesized, the amino acid chain is initially in a disordered, random state. In order to function correctly, the protein must fold into a specific, three-dimensional structure that is determined by the sequence of its amino acids. Protein folding is influenced by various factors, including the physical and chemical properties of the amino acids, the presence of other molecules, and the protein's environment.
From a biochemical perspective, protein folding is driven by the formation of weak, non-covalent interactions between different parts of the protein, such as hydrophobic interactions, hydrogen bonds, and electrostatic interactions. These interactions help stabilize the folded protein structure and give it the specific shape and function required for its role in the cell. Hydrophobic residues, which have nonpolar side chains that are not attracted to water, tend to be buried in the protein's interior during folding to minimize their unfavorable interactions with water. This burial of hydrophobic residues greatly limits the number of possible conformations the protein can assume and allows it to fold rapidly. Hydrogen bonding and electrostatic interactions can lead to the formation of secondary structure elements, such as alpha helices or beta sheets, which provide additional stability to the protein. The protein may also form tertiary structure elements, such as the packing of the interior and the fixation of surface loops.
From a physical science perspective, protein folding can be viewed as a thermodynamic process in which the protein molecule transitions from an unfolded state with high entropy to a folded state with lower entropy. This transition is driven by the minimization of the protein's free energy. The native state of a protein is typically the one with the lowest free energy and is stabilized by differences in entropy and enthalpy between the native state and the unfolded state.
Protein Structure Determination
While experimentally determined protein structures provide valuable information for drug design and discovery, the process of obtaining these structures can be time-consuming and resource-intensive. The preparation of pure, stable protein samples is an important step in the process, and the experimental techniques themselves, such as X-ray crystallography or NMR spectroscopy, can also be time-consuming. Once the structures have been determined, they are typically deposited in the Protein Data Bank (PDB) as coordinate files for future reference and use in research and development. Despite the challenges, the use of experimentally determined protein structures as a starting point for drug design and discovery has led to significant advances in the field.
Back To Menu
Traditional and Deep Learning Approaches in Protein Design
Traditional protein design seeks to maximize P(sequence|structure) by minimizing the energy of the target structure by Markov chain Monte Carlo (MCMC)-based search over side chain identities and conformations. The energy function is based on a combination of physical and statistical potentials, which are different types of energy terms that can be used to describe the behavior of the system. Physical potentials are based on physical laws and principles, such as the laws of motion and thermodynamics. They describe the energy of a system based on the positions, velocities, and other physical properties of the individual particles or atoms that make up the system. On the other hand, statistical potentials are based on statistical analysis of the properties of a large number of similar systems. They are used to approximate the energy of a particular system based on the probability that it will have certain properties or behavior. The capacity to generate diverse sequences and design multi-body interactions is a fundamental weakness of traditional protein design approaches. These restrictions have been lifted by deep learning approaches, which now provide enhanced design options.
Deep learning is a type of artificial intelligence that is built on two key principles: representation learning, and learning through local gradient descent using backpropagation. In representation learning, the algorithm adapts features to capture the regularities relevant to a specific task, regularities refer to patterns or structures in the data. These features can be organized into levels, with each level representing a different level of abstraction. The second principle, learning through local gradient descent, involves updating the parameters of the model in small steps based on the error between the predicted and actual output. This process is called backpropagation.
One challenge in deep learning is the "curse of dimensionality", which refers to the difficulty of learning generic functions in high-dimensional spaces. Given that the physical world has pre-defined regularities that arise from the underlying structure, exploiting these regularities, which often reflect the symmetries of a large system, can help to address the curse of dimensionality, which also forms the basis of most physical theories. The secret to breaking the curse lies in adapting neural networks to use low-dimensional geometry generated by physical measurements, for example, a grid in an image, a sequence in a time series, or position and structure in a molecule, and their associated symmetries, such as translations or rotations.
Before proceeding, it is worth noting that our work leverages the underlying principle of geometric regularity of protein structure to construct different representations, and those representations were evaluated on a state-of-the-art neural network architecture for protein design, called ProteinMPNN.
Back To Menu
